 rss
rss
Experiment: Conformational analysis of butane
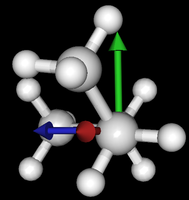
It's been quite a while since analysis_ethane, where I suggested that a look at butane might be interesting, too, as its structure of conformers is richer. As you can see by the following screenshots from MoleCuilder, there are four conformers.
| syn/eclipsed methyl | gauche | 120/eclipsed | anti |
 |  |  | 
|
However, the more complex an experiment, the more prone one is to making errors as well. I have made quite many: overwritten output files, stumbling over the limited precision of PDB files (use tremolo format if you're optimizing to very high precision), confusion when using the Python interface because of no "named arguments" support so far (contained in upcoming v1.6.1), copy&paste errors when building on the script I used for ethane.
But one thing especially impeded me: The quality of the optimization.
If one plans on investigating butane's rotational barriers, then one should have a fully annealed "anti" conformer state at hand.
But to make a long story short, here it is:
")
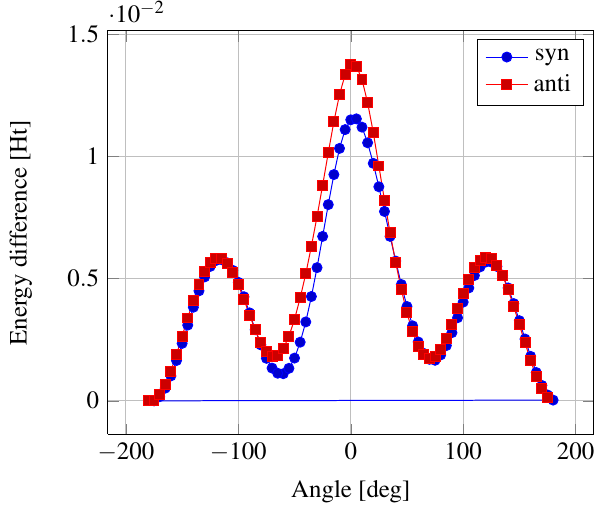
As you see in the conformer screenshots, I have done the rotation around the middle CC bond in the same manner as with ethane before. The conformer names relate to the following angles: syn (0°), gauche (60°), 120 (120°), anti (180°). I used two initial states: an optimized "syn" and an optimized "anti" state - both optimized to 1e-5 a.u. absolute error in energy in roughly 2000 steps. I have used 6-31G basis set and CLHF theory for optimization and rotation. If you'd like to reproduce the figure, using the docker image, both optimized conformer states are attached to this blog post including the update script to rotate around the CC bond and compute the energy. You need MoleCuilder version 1.6.1 which is soon to be released.
You notice that the two minima left and right of the central peak are not the same when starting from the "syn" state. They both correspond to the symmetric state of the same conformer. This is a clear indication that one needs to start from the lowest energy state, which is "anti". On the other hand, there is an energy difference in the central peak when starting from either state.
Comparing the above with experimental data at introorganicchemistry, you'll notice that the central peak is strongly overestimated by theory. However, a recent article by [Yirong Mo, JOCNote, 2012] states that experimental data and theoretical predictions do not match. This is why there are articles such as [Murcko, Castejon, Wiberg, 1996] and [Allinger, Fermann, Allen, Schaefer, JCP, 1997] calculating the energy differences to very high precision, estimating the syn/anti difference at around 0.087 Ht, while experimentally it is just 0.006 Ht, in chemical units 3.78 kcal/mole.
Here, we notice that the "syn" state is closer to the true energy difference as expected from theory, namely 0.0115 Ht. Note that [Yirong Mo, JOCNote, 2012] obtains roughly 0.01 Ht using Moeller-Plesset 2nd order and 6-31G(d) (with diffuse basis functions) where he optimized in each rotational step.
Having not optimized each step itself, I am satisfied with the results.
Attachments (8)
-
C4H10-rotational_barrier_anti_comparison-2017-09-10.png
(32.2 KB
) - added by 8 years ago.
Rotational barrier of Butane (C4H10)
-
C4H10_sequence2.png
(17.2 KB
) - added by 8 years ago.
C4H10 syn conformer
-
C4H10_sequence4.png
(15.4 KB
) - added by 8 years ago.
C4H10 120 conformer
-
C4H10_sequence5.png
(17.2 KB
) - added by 8 years ago.
C4H10 anti conformer
-
C4H10_sequence3.png
(21.6 KB
) - added by 8 years ago.
C4H10 gauche conformer
-
C4H10_anti_start.data
(899 bytes
) - added by 8 years ago.
Optimized C4H10 anti conformer
-
C4H10_start.data
(899 bytes
) - added by 8 years ago.
Optimized C4H10 syn conformer
-
C4H10_rotate-bond.py
(2.7 KB
) - added by 8 years ago.
python script using pyMoleCuilder for rotating butane around its central C-C bond
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Download all attachments as: .zip
Comments
No comments.